Дослідники зі США випробували на мишах експериментальну генну терапію спинальної м'язової атрофії (СМА), яка полягає у конвертації справного гена SMN2 у робочий варіант гена SMN1, який у пацієнтів із СМА є дефектним чи відсутнім. Такий підхід покращив моторні функції тварин і подовжив тривалість життя, особливо в разовій комбінації з наявним препаратом проти СМА. Результати опубліковані в журналі Science.
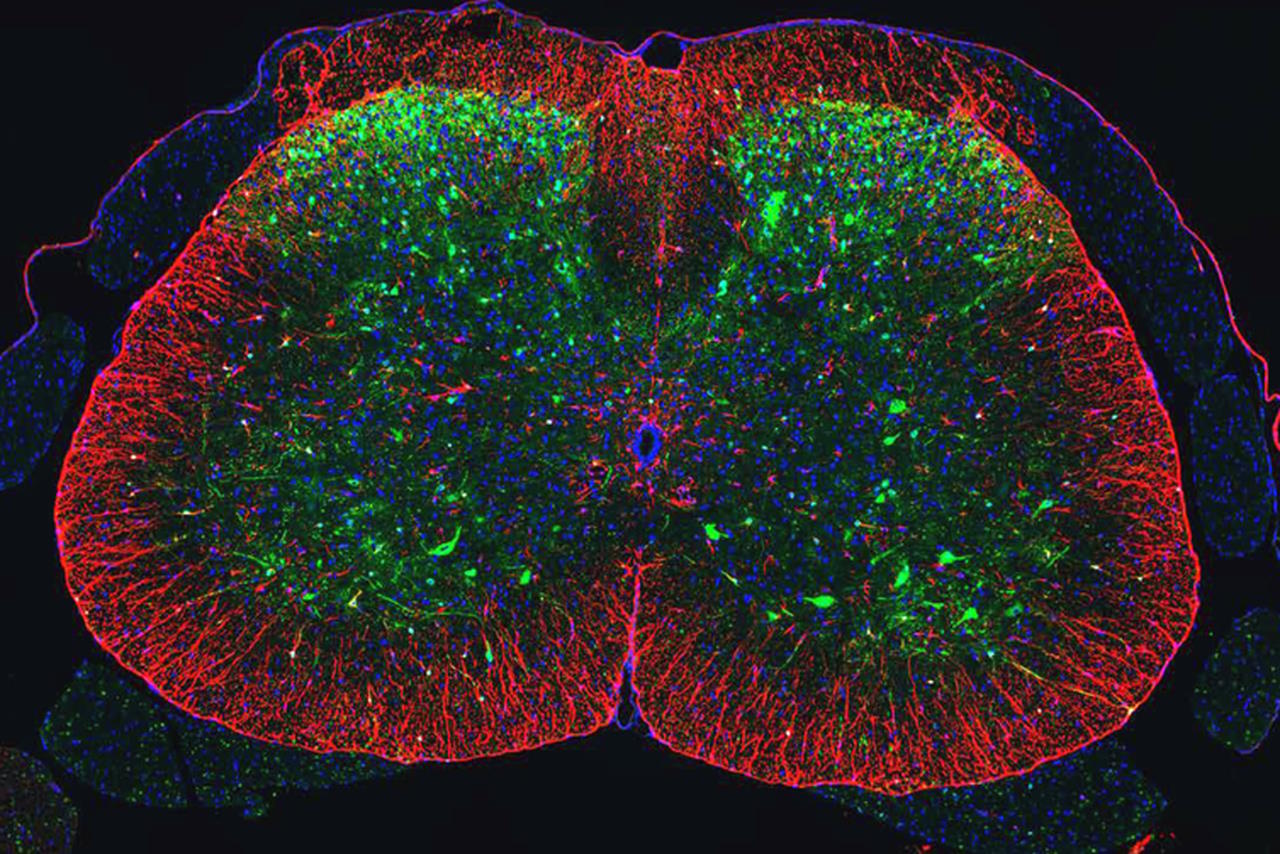
Поперечний зріз спинного мозку миші з СМА, де зеленим показано модифіковані вірусним вектором клітини. Dan Wang, Ailing Du / Broad Institute
Що не так з наявною генною терапією СМА?
Спинальна м'язова атрофія є важкою генетичною хворобою, яка спричиняється втратою або мутацією гена SMN1. У нормі білок, який він кодує, необхідний для роботи мотонейронів спинного мозку, а за його відсутності у хворих спостерігають атрофію м'язів з порушеннями рухової та дихальної функції. Це сильно погіршує якість життя пацієнтів, а у важких випадках призводить до смерті у перші роки життя.
Найефективнішим лікуванням захворювання наразі є генна терапія засобом Золгенсма, що доставляє до мотонейронів правильний варіант гена SMN1. Вона дозволяє спинити прогресування хвороби і зменшити її прояви, однак здатна спричиняти надмірну експресію гена в інших тканинах і органах, що може мати негативні наслідки. Окрім того, терапія оминає природну регуляцію утворення білка SMN і невідомо, як довго зберігатиметься її ефект, зважаючи на те, що сам геном не зазнає змін.
Яку альтернативу запропонували вчені?
Дослідники з Гарвардського університету з американськими колегами подумали, що ефективнішим підходом може бути невелика модифікація іншого гена — SMN2, який більш ніж на 99,9 відсотка збігається з геном SMN1. Він присутній у всіх пацієнтів з СМА, але його білок вкрай швидко деградує, що не дає йому змогу повноцінно замінити дефектний ген SMN1. Тож вчені модифікували п'ять ділянок гена SMN2 за допомогою технології редагування окремих азотистих основ, щоб збільшити експресію відповідного білка і зробити його стабільнішим у хворих на СМА. Простіше кажучи, перетворити SMN2 на SMN1.
Якого результату досягли модифікацією?
Науковці провели перші досліди на клітинах від мишей, у яких неробочий ген призвів до проявів СМА. Модифікація гена SMN2 збільшила рівень білка SMN у клітинах у 41 раз, повернувши його до нормального показника. Тому у наступних дослідах технологію випробували на хворих новонароджених мишах, яким редагування гена здійснили завдяки доставленню редакторів основ аденовірусами, що націлюються майже винятково на нейрони. Віруси доставляли за допомогою ін'єкції безпосередньо у спинномозкову рідину у дозі, що відповідна генній терапії наявним препаратом Золгенсма.
Вплинути вдалося на 43 відсотки мотонейронів, із яких 87 відсотків зазнали успішної зміни в гені. Підрахунок рухових одиниць скелетних м'язів на 12 день життя показав, що у лікованих експериментальною генною терапією мишей їхня кількість складає 91 відсоток від кількості рухових одиниць у здорових тварин. Натомість у нелікованих тварин або лікованих Золгенсмою збереглося за той час лише близько 50 відсотків рухових одиниць. З редагуванням основ у мишей була краща ще й реакція м'язів на стимуляцію мотонейронів. Власне, вона не відрізнялася від показників здорових тварин або лікованих Золгенсмою. Генна терапія також подовжила тривалість життя піддослідних тварин на 33 відсотки, з 17 до 23 днів.
Та досліди показали, що можна досягти ще кращих результатів одноразовою комбінацією експериментальної генної терапії та препарату проти СМА нурсінерсену, який зазвичай використовують регулярно протягом життя. У такому разі моторні функції, координація рухів та м'язова сила майже не відрізнялися від показників здорових тварин, а тривалість життя мишей зросла до 111 днів. Науковці сподіваються, що запропонована ними терапія спрацює так само добре і в людей, покращивши якість та тривалість життя пацієнтів з СМА.



