Управління з продовольства та медикаментів США (FDA) прискорено схвалило генну терапію препаратом «Елевідіс» (Elevidys) проти м'язової дистрофії Дюшена — важкого генетичного захворювання, яке характеризується втратою функціональності м'язів. Оцінювання ефективності лікування ще триває, але попередні випробування свідчать, що засіб збільшує у пацієнтів рівень білка, який може зменшити прогресування хвороби. Рішення оприлюднене на сайті FDA.
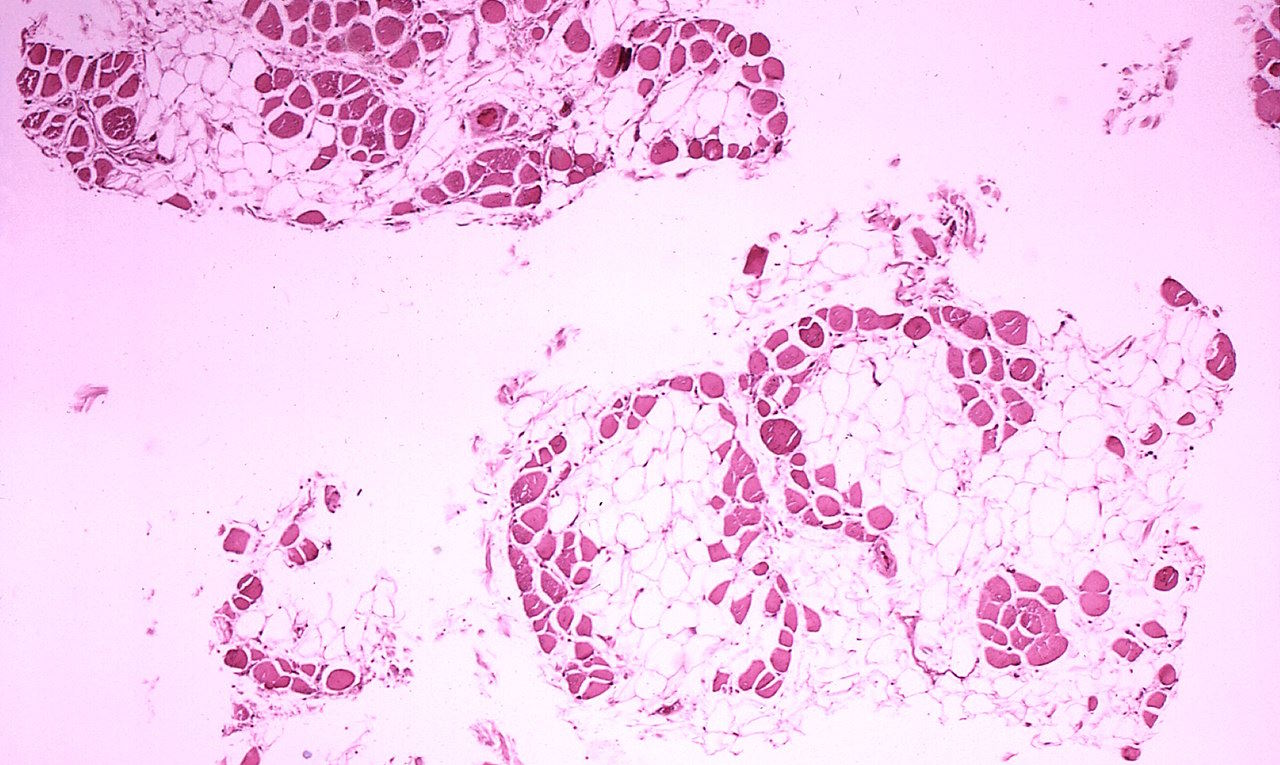
Зразок гістологічного дослідження м'яза людини із дистрофією Дюшена, на якому помітно заміщення м'язових клітин жировими. Dr. Edwin P. Ewing, Jr. / PD-USGov-HHS-CDC / Wikimedia Commons
Що це за хвороба?
М'язовою дистрофією Дюшена називають спадкову хворобу, спричинену мутацією в гені дистрофіну, який відіграє ключову роль у підтриманні цілісності м'язових клітин. Пацієнти з цим діагнозом є майже завжди чоловіками, які із раннього дитинства страждають від м'язової слабкості, через яку у них розвивається складність із ходьбою, утриманням постави, а при прогресуванні хвороби — і з диханням та роботою серця. Захворювання уражає приблизно одного із 3 300 хлопчиків, а ефективного лікування розладу наразі немає. Але як і з багатьма іншими генетичними хворобами, великі надії покладають на генну терапію.
Як діє генна терапія проти хвороби?
Щойносхвалений FDA «Елевідіс» від німецької фармкомпанії Sarepta Therapeutics при внутрішньовенній ін'єкції вводить в організм модифіковані аденоасоційовані віруси, що доставляють у клітини ген мікродистрофіну. Це вкорочена версія природного великого дистрофіну, дефіцит якого він покликаний перекрити. Випробування показали, що засіб підвищує у дітей рівень мікродистрофіну до рівня, який очікувано покращить стан пацієнтів, без надмірних ризиків здоров'ю. Цього достатньо для процедури прискореного схвалення ліків, що її застосовують до небезпечних хвороб без наявного лікування.
Наразі «Елевідіс» дозволили використовувати у пацієнтів без протипоказань до нього віком від 4 до 5 років, коли починає проявлятися м'язова дистрофія Дюшена. Однак управління очікує на результати випробування препарату щодо його клінічної ефективності, на основі якого може переглянути рішення про дозвіл.



